- support
- info@evidentic.com
- +49 (0) 30 959 99 8831

Antibody-drug conjugates (ADC), a new emerging class of biologics, have upgraded the therapeutic index of biological interventions and chemotherapy. Almost a century ago, German physician and scientist Paul Ehrlich coined the concept of antibody-drug conjugate and suggested the attachment of toxins to the antibodies to focus on their therapeutic specificity. Later, as a part of the evolution, in the 1980s and 1990s, monoclonal antibody-drug conjugates based on mouse IgG molecules and chimeric/humanized mAbs were developed.
The antibody-drug conjugate platform has mainly facilitated a broader therapeutic window in the treatment of cancer. The features of antibody-drug conjugates enhance the exclusive delivery of highly potent drugs to tumor cells while excluding healthy cells, mitigating off-target toxicities, the main clinical obstacle of chemotherapy and immunotherapy.
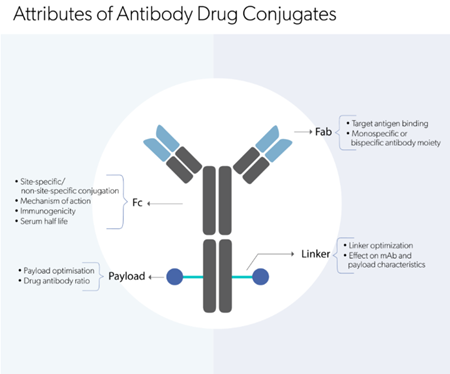
Antibody-drug conjugate technology comprises three major components:
The right choice of antigen as the target is critical in the development of monoclonal antibody (mAb) component in ADC against cancer. Criteria for the selection of unique antigen targets are:
The most commonly targeted antigens in antibody-drug conjugate development include ERBB2, CD19, CD33, CD22, and MSLN (mesothelin).
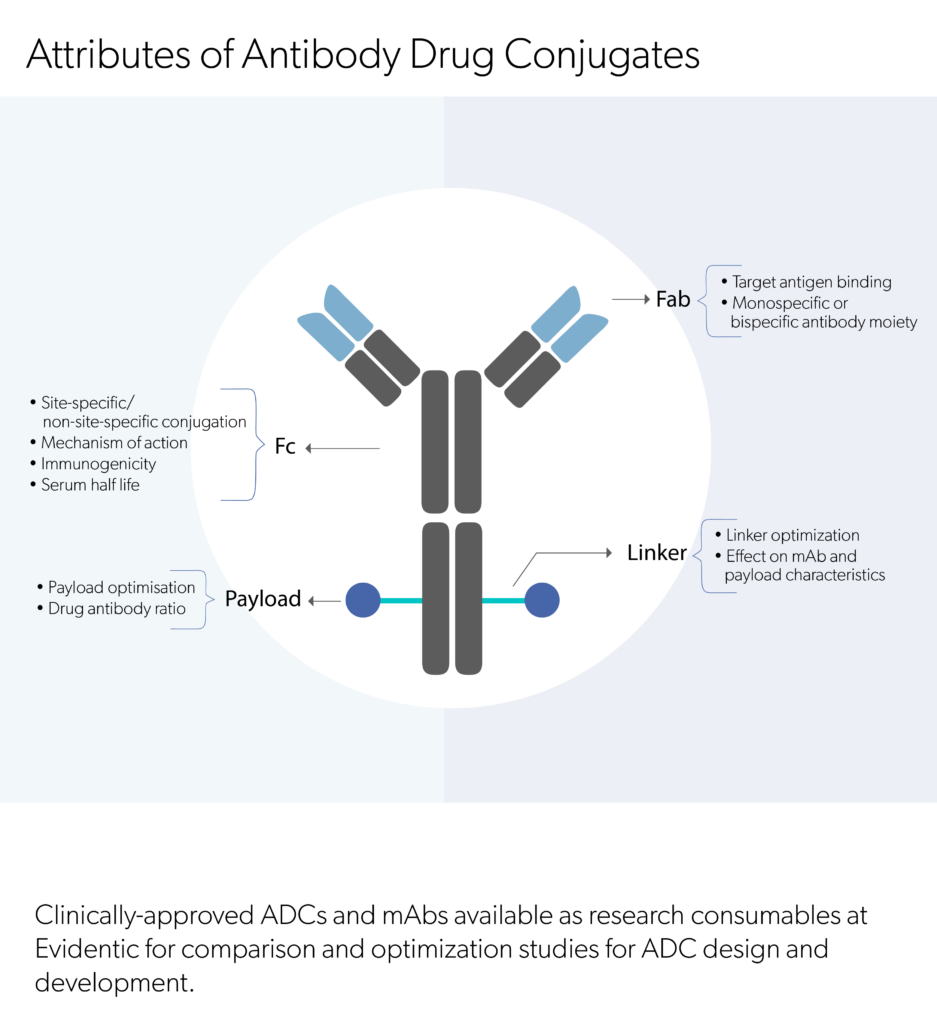
The accurate selection of monoclonal antibody(mAb) is vital in the antibody-drug conjugate design. The factors to consider while determining the monoclonal antibody (mAb) includes:
Upgradation of antibodies in different generations of ADC started from a murine antibody to a mouse/humanized chimeric antibody, humanized monoclonal antibody(mAb), and finally to a fully human monoclonal antibody(mAb). The latest update with fully human monoclonal antibodies has helped overcome immune response and production of anti-human antibodies. Recently, an ADC was designed using a fully human anti-Guanylyl Cyclase C (GCC) mAb conjugated to a highly cytotoxic drug monomethyl auristatin E (MMAE) through a protease-cleavable peptide linker demonstrating promising antitumor activity in GCC-expressing cells both in vitro and in vivo. Recently, the use of bispecific antibodies, antibody fragments, and other formats are being explored for antibody-drug conjugation. Several preclinical evaluation studies show a better efficacy for bispecific-based ADCs.
Read more: Bispecific antibodies
Linker selection and optimization are some of the key deciding factors in ADC therapeutic outcomes. The linker must be stable enough to resist any systemic condition that might cause proteolytic degradation. Thus, it must ensure that cytotoxic drug is not released before reaching the target site to avoid escape into blood-stream and consequent off-target cytotoxicities. Another factor to be considered while selecting a linker is the effect of linker conjugation on antigen-binding and payload-drug efficacy. Two types of linkers are generally employed in ADC structure: Non-cleavable and Cleavable linker.
Non-cleavable linker
Noncleavable linkers, such as non-reducible maleimidocaproyl (MC), provide high stability to the conjugate structure. A successful commercial example is the 4-maleimidomethyl cyclohexane-1-carboxylate (MCC) used in Kadcyla®. With non-cleavable linkers, the ADC degrades only upon internalization within the lysosome, killing the target cell. The advantage here is the apparent minimization of off-target effects. Moreover, certain non-cleavable linkers have shown to improve target-binding affinity and drug efficacy. For example, trastuzumab-SMCC-DM1 demonstrated better efficacy rates when compared to trastuzumab alone or on conjugation with a reducible linker. In addition, ADCs conjugated to drugs with charged amino-acids via non-cleavable linkers make good options for bystander effect as they show low membrane permeability. Alternatively, MC linkers can also be used as spacers to separate mAb and the cleavable linker.
Cleavable linkers
The majority of the approved ADCs employ cleavable linkers. Such linkers exploit the differences in the target extracellular or intracellular microenvironmental conditions. For example, change in pH or the presence of certain enzymes in the tumor microenvironment. The different classes of cleavable linkers include:
In general, enzyme/protease-sensitive linkers offer better stability and selectivity than acid-sensitive or disulfide linkers.
Payloads are cytotoxic compounds that are highly potent and too toxic to be used as small-molecule drugs for chemotherapy. Hence, by conjugating them to mAbs, targeted delivery of these drugs can be achieved. Payloads used for ADCs must possess (i) high stability in circulation and lysosome, (ii) cytotoxic activity in sub-nanomolar range, (iii) solubility in aqueous environment, (iv) low immunogenicity, and (v) long half-life. The current generation of commercial ADCs and ADCs under development employs the following classes of payloads.
2. DNA-damaging agents: These compounds bind to DNA and disrupt DNA replication. Examples include:
3. RNA-polymerase inhibitors: Amatoxins, spliceostatin C and thailanstatins are RNA polymerase II inhibitors being explored as payloads for ADCs. They disrupt transcription and protein synthesis. Examples include α-amanitin and β-amanitin, which are being investigated in several preclinical candidates.
The clinical efficacy of the ADC is dictated by linker stability, ADC internalization and adequate tumor cytotoxicity. To achieve this, the number of drug molecules to be conjugated to each mAb, referred to as the drug-antibody ratio, must be optimized. Generally, the DAR ranges from 2-8 drug molecules per antibody. In this context, the hydrophobicity of the ADC should also be taken into consideration. An increased hydrophobicity via a hydrophobic linker and hydrophobic payload can lead to ADC aggregation and clearance. Another challenging aspect is to achieve homogenously conjugated ADC drug molecules with identical pharmacological properties. The possible distribution of payloads across different potential conjugation sites can also lead to heterogeneity. ADC DAR and homogeneity are currently active areas of research to minimize batch variability.
Designing ADCs? Evidentic is your one-stop shop for biotherapeutic reference molecules for research.
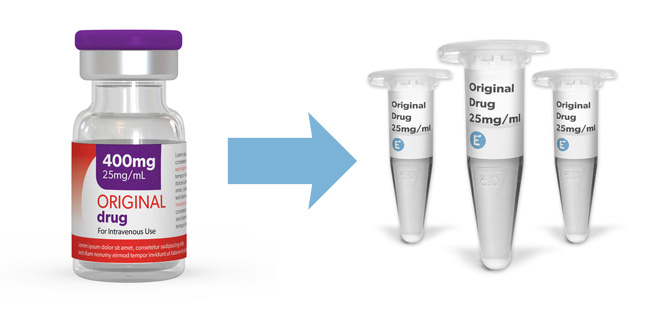
Purchase therapeutic ADCs and other biologics as aliquots, in small quantities. All the molecules have been repackaged from the original medicines, retaining their original formulation (concentration and buffer). You have access to EMA approved therapeutic molecules for your early drug development programs as research consumables.