- support
- info@evidentic.com
- +49 (0) 30 959 99 8831

With a view to making these lifesaving products globally affordable and accessible, several “biosimilar mAbs” are being developed. These biosimilar monoclonal antibodies are highly similar in quality, safety, and efficacy to the original mAbs and express no clinically significant differences to the existing products. However, they can be different with respect to the inactive components present in the original drug and the method of production.
Licensure of biosimilars can only be obtained after the evaluation of data from a quality, nonclinical, and clinical perspective. So as to achieve this, the role of the originally licensed drug, oftentimes called the “reference drug”, comes into play. The attributes of biosimilars must comply with the demonstration of quality, efficacy, and safety as exhibited by the reference drugs, for the regulatory approval of biosimilar drugs. Relevant, robust, and reliable assays and methods are crucial in determining the eventual success of biosimilar approval.
The similarity of biosimilar and reference mAbs must be assessed in sequential order. The comparability exercise implements a fit-for-purpose approach to evaluate deviations in structural, functional, and biological activity.
Since the manufacturing process of the reference drug is often undisclosed, the first step in biosimilar development is to investigate the reference mAb in detail and then establish the manufacturing method for the biosimilar. To accomplish this, first and foremost, the quality target product profile (QTPP) needs to be identified which involves analyzing multiple batches of the reference drug and characterizing its quality attributes. The development of QTPP requires an extensive evaluation of the structural and physico-chemical properties such as terminal amino acid variants, charge variants, and oligosaccharide profiles. The characteristics of the product, are classified according to their criticality to determine the critical quality attributes (CQAs) that might impact the bio-similarity, as well as clinical safety and efficacy.
The manufacturing process of the biosimilar mAb is optimized based on the QTPP, to achieve quality characteristics highly similar to that of the reference mAb. Selection of production methods like expression system, cell culture conditions and purification procedure can impact the mAb structure (for example, glycosylation patterns) and impurity profiles. This in turn can affect the physico-chemical and biological properties. Therefore, a wide range of analytical and functional assays are employed to examine the structural heterogeneity and to demonstrate the mechanism of action of the newly formulated biosimilar.

The comparative studies begin with in-vitro analysis and then depending on the upshot, if necessary, in-vivo studies are conducted. The non-clinical in-vitro programme focuses on the quality comparability of the biosimilar and the reference drug. Several analytical methods such as mass spectrometry, surface plasmon resonance (SPR), ELISA and flow-cytometry are used to compare the quality attributes including,
Furthermore, functional assays are performed to compare and evaluate the mechanism of action, viz. antibody-dependent cellular cytotoxicity (ADCC) or complement-dependent cytotoxicity(CDC); and also to detect differences in concentration–activity relationship. Overall, these studies must be specific and sensitive enough to conclude that the observed differences will not influence clinical performance.

If the biosimilar quality-comparability in vitro studies result in debatable satisfaction, and identification of issues that can hinder direct human use, an in-vivo animal study is considered necessary. Animal studies are performed on relevant species to understand the pharmacokinetics (PK), pharmacodynamics (PD), safety and immunogenicity. In-vivo experiments focus on comparing the dose-response curve and dose-toxicity in diseased animal models. Assays are used to evaluate the functional effects on relevant PD biomarkers and to detect anti-drug antibodies.
PD studies involve single-plex or multi-plex assays that can measure biomarkers such as mRNA level, protein expression and localization in order to confirm the drug effect on the target and clinical outcomes. The PD markers selected should be sensitive enough to measure and compare the dose–response relationships in accordance with the reference drug.
To ensure a better comparison of clinical performance, a randomized, double-blinded study is conducted in a homogenous patient population (for example, same stage of disease severity and/or same line of treatment). The clinical data available for the reference drug determines the selection of study population and study design. The safety data of the reference drug is also used to design assays, such as ELISA, to detect adverse immune response in the patients. The assay should be compatible to assess immunogenicity of both the reference mAb and biosimilar mAb.
If the reference drug is used for more than one disease indication (for example, immunomodulatory mAbs), approval of its biosimilar will require individual validation in each case. It is important to evaluate the mechanism of action and immunogenicity as it may vary in different clinical settings. Furthermore, from a pharmacovigilance standpoint, immunogenicity diagnostic assays need to be designed for every biosimilar. These assays can be used in the post-marketing phase to assess risk management in a more diverse patient population.
Hence, we can culminate that bio-therapeutic products have shown great potential in treating many fatal and chronic disease conditions. Some of these targeted therapy products are already well established leading pharmaceutical revenue sources. As the validity of the market exclusivity period on certain mAb products have expired, or are nearing expiry, considerable attention has turned towards producing similar bio-therapeutic products. This necessitates a systematic analysis of the quality, efficacy, and safety of the biosimilar to the earlier approved reference mAb and emphasizes its role in the development of comparability assays.
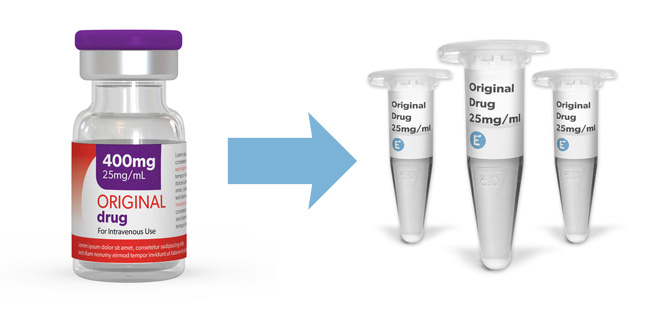
Having access to clinical-grade molecules from licenced drugs is essential to performing early drug development programs. At Evidentic you have access to these therapeutic molecules in small and convenient aliquots, including different manufacturing batches. Browse our portfolio and order today your desired molecule.