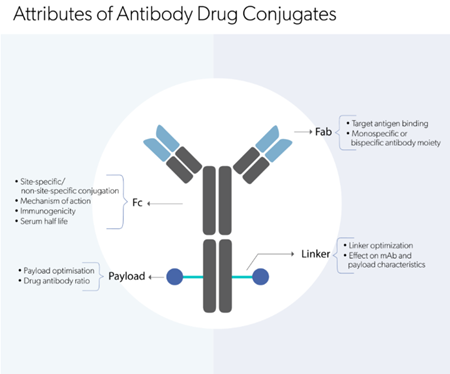
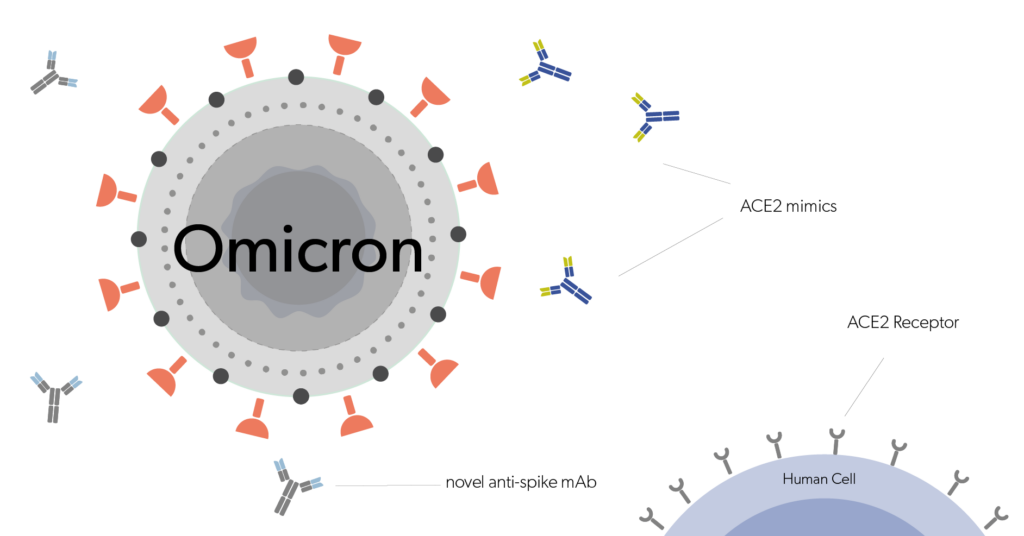
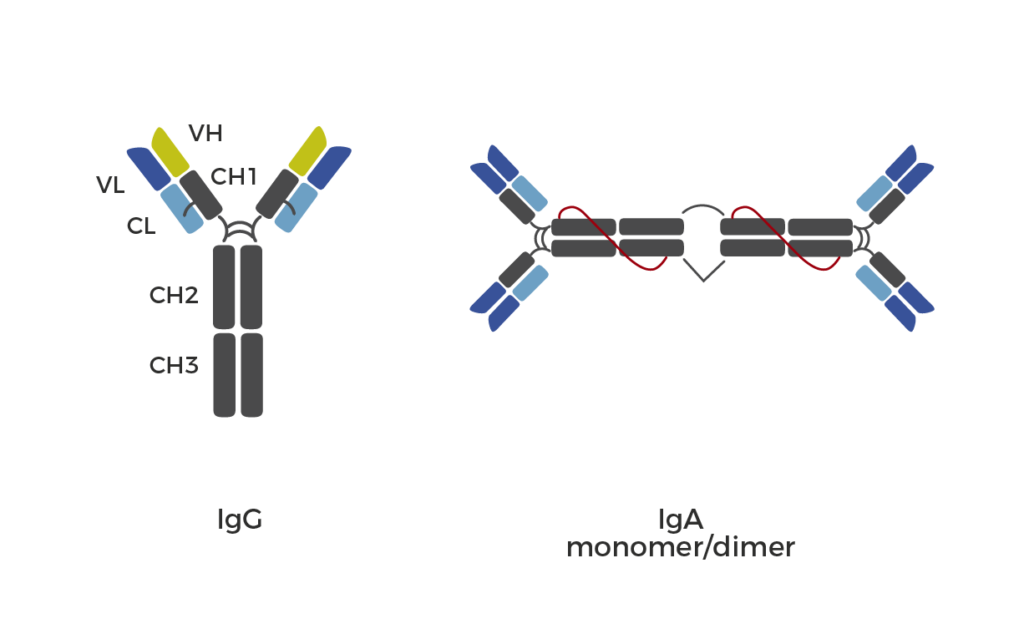
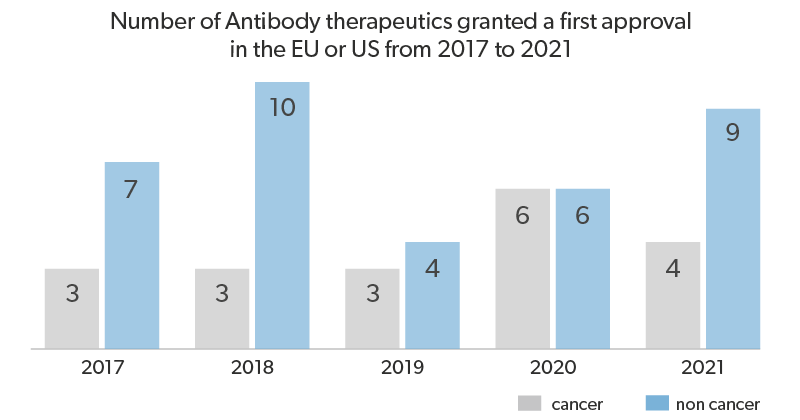- support
- info@evidentic.com
- +49 (0) 30 959 99 8831

Orphan drugs are designed to treat disease conditions that are rarely observed in people. Rare diseases manifest only in 6-8% of the world population; hence such conditions are defined as orphan diseases. However, globally, the prevalence of rare diseases is seen to rise in the last couple of years. In the US, the Orphan Drug Act (ODA) came into existence in 1983 to facilitate research and development of products that exhibit promise for the diagnosis and/or treatment of orphan diseases. The FDA’s Office of Orphan Products Development (OOPD) is responsible for orphan drug development and authorization. Whereas, in the EU, the Committee for Orphan Medicinal Products (COMP) is the European Medicines Agency’s (EMA) committee responsible for recommending orphan designation of medicines for rare diseases. Both regulatory bodies authorize financial incentives to attract industry’s interest, market exclusivity and waive off tax for research and development-related expenses. Based on drug type, orphan drugs are categorized into biological and non-biological drugs. Since biologics elicit fewer side effects in the treatment; therefore, approval of biological orphan drugs for multiple indications steers the growth of the orphan drugs market.
Interestingly, the sales forecast of orphan drugs estimates a growth of 12.3% per year, reaching $242bn in 2024. And with respect to the overall prescription drug market, orphan drugs anticipated sales account for a little over a fifth of all prescription drug sales by 2024.
Firstly, in order to acquire Orphan Drug Designation (ODD), the drug or biologics should satisfy three major criteria as follows
Importantly, ODD is applicable to both the active moiety and the condition but may be exempted for the product formulation. Also, derivatives of a given active moiety such as esters, salts, and other non-covalent categorize as the same drug when it comes to ODD.
In the US, the process is initiated when ODD requests are submitted to the FDA’s OOPD. In order to obtain the ODD status for the targeted drug and condition, submissions can be made at any time during drug development before New Drug Application (NDA) or biologics license application (BLA). A sponsor is not obliged to open an investigational new drug (IND) to request the ODD status since an ODD request is not submitted as an amendment to an IND. Orphan Drug Modernization Plan initiated in 2017 by the FDA facilitated responding to all new ODD requests within 90 days of their receipt.
Similar to FDA’s examination timeline, COMP assessment procedure includes 90 days from validation. As a part of the evaluation in the EU, companies categorized as micro, small, or medium-sized enterprises can benefit from further incentives. If COMP recommends issuing a license, the exclusivity period for orphan drugs marketed within the EU is ten years. The COMP also advises and assists the European Commission on matters related to orphan medicines, including:

In the case of both regulatory bodies, COMP and OOPD, the sponsor must submit orphan annual reports summarizing the status of development of the desired orphan drug or biologics every year until the marketing application is approved.
The lack of or inefficient legal and policy frameworks in many countries make it challenging to access therapies for rare diseases. High cost for the therapies and insufficient funding is also the other factors contributing to the limited access and affordability of orphan drugs. However, such obstacles can be overcome through education towards payers, responsible and evidence-based pricing, and innovative contracting. Addressing these issues, especially in middle-income and emerging markets, will help contribute to the growth in rare disease therapeutic areas.
The classification of orphan drugs may vary from country to country. Various regulatory authorities take different approaches to classify rare diseases. For example, the US- FDA mainly categorize an orphan disease based on the disease prevalence, that is, simply the number of people suffering from the disease. In comparison, the EMA calculates the prevalence proportion, which considers the proportion of people in the population that have the disease. The table summarizes some of the biologic drugs among various therapeutic areas that have been granted a European orphan designation according to Regulation (EC) No 141/2000 (updated on March 2021).
In addition, the EMA also mentions a list of drugs that have a European marketing authorization (MA) for one or more indications of use for a rare disease but have either not been granted a European orphan designation or the designation was withdrawn. These drugs may or may not have an ODD in other countries. For example, Pembrolizumab (Keytruda®) is an orphan drug in the US but does not hold ODD status in the EU. The US FDA has conferred an orphan drug designation for Keytruda for three separate indications for malignant melanoma from stage IIB through IV.
| Therapeutic areas | International Non-proprietary name (INN) | Molecular class | Target /MoA | Therapeutic indications |
| Oncology | Brentuximab vedotin | Antibody-drug conjugate | CD30 directed delivery of the cytotoxic agent | CD30-positive Hodgkin lymphoma (HL), systemic anaplastic large cell lymphoma (salcl) |
| Inotuzumab ozogamicin | Antibody-drug conjugate | Binds CD22 mediating ADC-CD22 internalization in tumor cells | CD22-positive B cell precursor acute lymphoblastic leukaemia (ALL) | |
| Blinatumomab | Bispecific | T cell engager that binds to CD19 on B-lineage cells and CD3 on T-cells | Refractory B -precursor acute lymphoblastic leukaemia (ALL) | |
| Obinutuzumab | Monoclonal antibody | Targets CD20+ B-cells and induces ADCC | Chronic lymphocytic leukaemia (CLL) advanced follicular lymphoma |
|
| Moxetumomab pasudotox | Antibody-drug conjugate | CD22-targeted immunotoxin delivery | Hairy cell leukaemia (HCL) | |
| Gemtuzumab ozogamicin | Antibody-drug conjugate | CD33 directed delivery of cytotoxin | CD33-positive acute myeloid leukaemia (AML) | |
| Polatuzumab vedotin | Antibody-drug conjugate | CD79b-targeted delivery of anti-mitotic agent |
Relapsed/refractory diffuse large B-cell lymphoma (DLBCL) | |
| Mogamulizumab | Monoclonal antibody | Binds to CCR4 resulting in depletion of the target cells |
Mycosis fungoides (MF) or Sézary syndrome (SS) | |
| Dinutuximab beta | Monoclonal antibody | Targets disialoganglioside 2 (GD2) | High-risk neuroblastoma | |
| Siltuximab | Monoclonal antibody | Binds to IL-6 preventing IL-6 mediated signalling | Multicentric Castleman’s disease (MCD) | |
| Autoimmune diseases | Belantamab mafodotin | Antibody-drug conjugate | Targets B cell maturation antigen (BCMA) on tumor cells, leading to internalization | Multiple myeloma |
| Daratumumab | Monoclonal antibody | Binds to CD38 blocking CD38 mediated signalling | Multiple myeloma | |
| Hematology | Eftrenonacog alfa | Recombinant protein | Recombinant human coagulation factor IX | Haemophilia B |
| Crizanlizumab | Monoclonal antibody | Binds to P-selectin blocking P-selectin/PSGL-1 interaction | Sickle cell disease | |
| Caplacizumab | Antibody fragment | Targets von Willebrand factor inhibiting its interaction with platelets |
Acquired thrombotic thrombocytopenic purpura (attp) thrombosis |
|
| Albutrepenonacog alfa | Fusion protein | Recombinant human coagulation factor IX | Haemophilia B | |
| Luspatercept | Fusion protein | Binds to selected transforming growth factor-β (TGF-β) superfamily ligands |
Transfusion-dependent anaemia associated eith low and intermediate-risk myelodysplastic syndromes (MDS) and beta thalassaemia. |
|
| Eculizumab | Monoclonal antibody | Binds to the complement protein C5 | Atypical haemolytic uraemic syndrome (ahus) generalized myasthenia gravis (gmg) neuromyelitis optica spectrum disorder (NMOSD) |
|
| Lanadelumab | Monoclonal antibody | Targets and inhibits active plasma kallikrein |
Hereditary angioedema (HAE) | |
| Metabolic disorders and Neurology | Cerliponase alfa | Recombinant protein | Recombinant human enzyme tripeptidyl peptidase-1 (rhTPP1) | Neuronal ceroid lipofuscinosis type 2 (CLN2) disease |
| Sebelipase alfa | Recombinant protein | recombinant human enzyme lysosomal acid lipase (rhLAL) | Lysosomal acid lipase (LAL) deficiency |
|
| Velmanase alfa | Recombinant protein | Recombinant form of human enzyme alpha-mannosidase | Alpha mannosidosis | |
| Vestronidase alfa | Recombinant protein | Recombinant human beta-glucuronidase (rhGUS) | Mucopolysaccharidosis VII (MPS VII; Sly syndrome | |
| Parathyroid hormone | Recombinant protein | Recombinant human parathyroid hormone | Chronic hypoparathyroidism | |
| Cenegermin | Recombinant protein | Recombinant form of human nerve growth factor | Neurotrophic keratitis |
|
| Pegvaliase | Recombinant protein | Recombinant Anabaena variabilis phenylalanine ammonia lyase | Phenylketonuria (PKU) | |
| Somapacitan | Fusion protein | Recombinant human growth hormone derivative with albumin binding moiety | Adult growth hormone deficiency (AGHD) | |
| Asfotase alfa | Fusion protein | Recombinant tissue-nonspecific alkaline phosphatase-Fc-deca-aspartate fusion protein with enzymatic activity |
Paediatric-onset hypophosphatasia | |
| Elosulfase alfa | Recombinant protein | Recombinant form of human N-acetylgalactosamine-6-sulfatase (rhGALNS) | Mucopolysaccharidosis, type IVA (Morquio A Syndrome, MPS IVA | |
| Velaglucerase alfa | Recombinant protein | Recombinant form of glucocerebrosidase | Type 1 Gaucher disease. |
|
| Bone diseases | Burosumab | Monoclonal antibody | Binds to fibroblast growth factor 23 (FGF23) leading to inactivation | X-linked hypophosphataemia |
| Infectious diseases | Obiltoxaximab | Monoclonal antibody | Binds to protective antigen (PA) of B. anthracis | Inhalational anthrax |
|
Reference: Orphanet Report Series – Lists of medicinal products for rare diseases in Europe. March 2021 http://www.orpha.net/orphacom/cahiers/docs/GB/list_of_orphan_drugs_in_europe.pdf
|
||||
Doing research on rare diseases? Biologic drugs with high specificity have enabled treatment for several disease conditions that were previously thought to be undruggable. Hence biologic drugs are steering the orphan drug market. Buy aliquots of EMA and FDA orphan-designated biologic drugs for research use.
Check out the list of all biologic drugs here.