- support
- info@evidentic.com
- +49 (0) 30 959 99 8831

Biologics or recombinant protein drugs, such as therapeutic antibodies and recombinant hormones and enzymes, require an expression system for their production. As a result, manufacturing and purification require continuous monitoring of process sterility and tight microbial control strategies. In order to obtain a sterile and patient-safe biologic drug, the manufacturing process must ensure that the final product is free from intrinsic impurities (upstream) and extrinsic contaminants (downstream). These include:
Endotoxins are amphiphilic lipopolysaccharides (LPS) located in the outer cell membrane of gram-negative bacteria. Endotoxins are categorized as pyrogens (fever inducing foreign substances) since they trigger TLR response, leading to inflammation, fever, and sometimes shock and organ failure. Hence, pyrogenic contamination of biological drugs and the excipients used to formulate them are a concern for biologics manufacturers. At the time of submission of an investigational new drug (IND) or biologics license application (BLA), it is important to include controls on pyrogens, including endotoxins, as a part of the chemistry, manufacturing, and control information.
LPS or bacterial endotoxin comprise three main parts: antigen, core oligosaccharide, and the Lipid-A molecule, which is the conserved and active toxic component of bacterial endotoxin. If the bacterial cell wall is lysed after it reaches the human bloodstream, Lipid-A molecule of the bacterial endotoxin activates the toll-like receptor 4 (TLR4) of the immune system, releasing different types of vasoactive peptides and cytokine mediators responsible for cell apoptosis and fever, diarrhoea, vomiting, and also fatal endotoxic shock (septic shock). Therefore, bacterial endotoxin/LPS characteristics and structural aspects have been utilized for the development of endotoxin detection tests and removal methods. Hence, endotoxin detection in biologics is a necessity to ensure that the therapeutic drugs are endotoxin-free.
It is always better to design manufacturing processes that produce endotoxin-free end products, rather than include steps later on to remove endotoxins in the final product. From an FDA perspective, in order to implement Quality by Design manufacturing concepts, the endotoxins testing strategies must be tailored depending upon the specific biologic product and process development. Additionally, appropriate risk management steps are required to assure consistent final product quality. Suitable in-process testing methods should be employed to monitor the production process areas that pose a risk of endotoxins contamination.
Conventionally, the bacterial endotoxins test (BET) and the rabbit pyrogen test (RPT) have been used to detect endotoxin levels during biologic drug development.
Recently, the low endotoxin recovery (LER) phenomenon has been described for LAL tests. LER is the inability to recover known amounts of purified endotoxin from biological formulations, and can be caused by the presence of chelating agents and surfactants in biological formulations. The LER phenomenon can be evaluated using suitable reference standard endotoxin, control standard endotoxin, and naturally occurring endotoxin.
In general, endotoxin acceptance criteria are set such that the endotoxin administration level does not exceed the pyrogenic threshold. Thus, the mode of administration is also an important determinative factor. In the case of intravenous and subcutaneous administration, the upper limit is set at 5 EU/Kg/hour or 100 EU/m2/hour. And for intrathecal, the threshold is set at 0.2 EU/kg/hour. These values denote the maximum combined endotoxin exposure level from all agents (including concomitantly administered drugs), provided there is no clinically significant increase in body temperature at these exposures. For lyophilized drugs, the endotoxin contribution from the diluent or reconstitution fluid is to be considered while setting the acceptance threshold.
Importantly, for the development of combination therapies, the combined endotoxin levels of all drugs must be evaluated. Combination therapy involves the administration of two or more biologics to obtain a synergistic clinical effect. Most of these drugs are originally designed as monotherapies, therefore the individual impurity levels and mode of administration must be taken into consideration. In the case where EU exceeds the recommended levels, phase-wise drug administration could be an alternative treatment option.

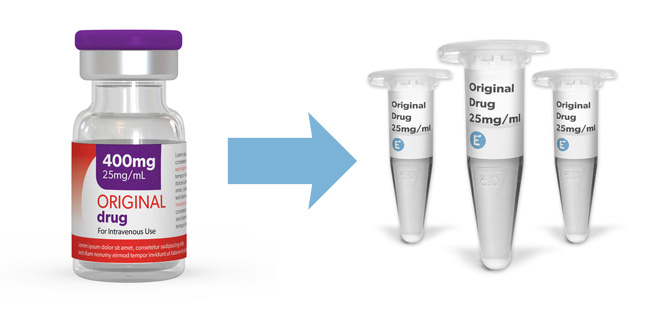
Original commercial drugs contain tested and approved endotoxin levels, therefore they can be used as reference standards for R&D purposes. If you need clinical-grade molecules for your early drug development programs, our drug aliquots are the perfect fit for you. Your advantages are: