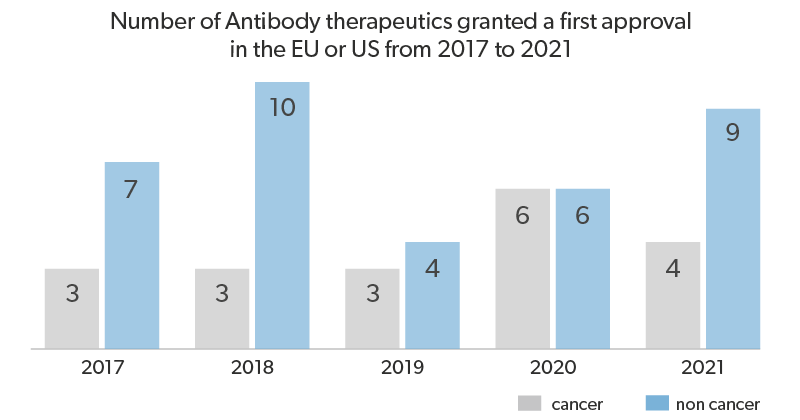- support
- info@evidentic.com
- +49 (0) 30 959 99 8831

Biological products are stratified as diverse and complex molecules, unlike conventional small molecule drugs. This intrinsic heterogeneity poses a challenge to characterize biological medicines, underlining the need for a reference standard. A reference standard, simply translates as a formally approved biologic molecule whose biological and therapeutic effects are well established.
Manufacturers use reference standards to provide solutions for different purposes such as discovery, regulatory compliance, and process efficiency.
Different and unique purposes of reference standards vary from (a) assigning a potency value to a biologic drug, (b) confirming identity of a drug substance, (c) determining the performance of quality control (QC) assays or suitability tests, (d) testing the presence of specific product and process related impurities, (e) determining molecular weight distribution and weight- average molecular weight, and (f) assessing qualification and validation of analytical and production methods. The comparator approved types of reference standards can be sourced commercially, if available, or generated in-house.
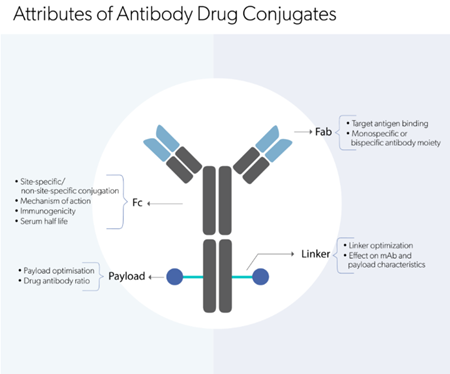
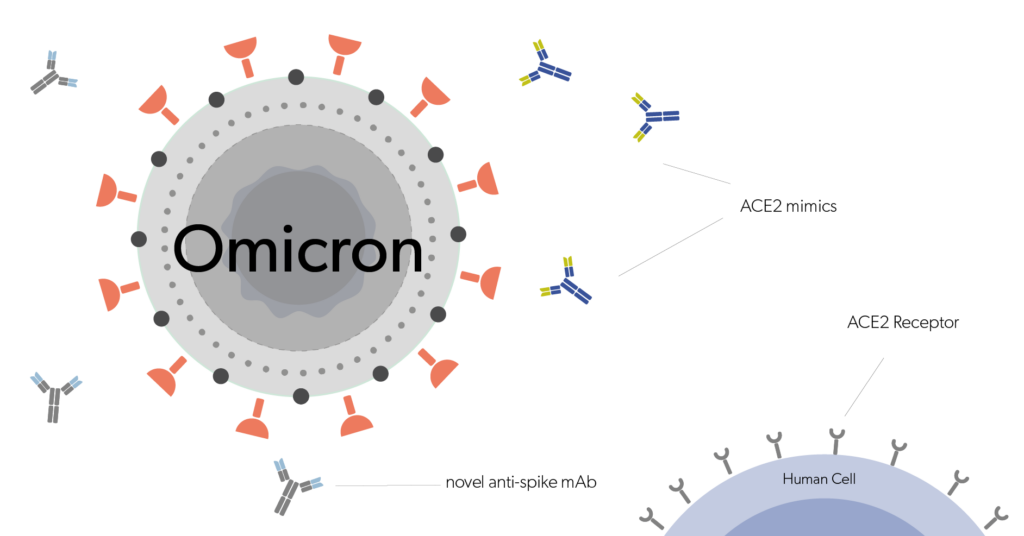
Among biologics, biotherapeutic monoclonal antibodies (mAbs) have had a major impact on healthcare, especially in the fields of oncology and inflammation. Most of the approved therapeutic mAbs are of the IgG-class antibody, which is the common type of antibody seen in blood. In comparability exercise, the assessment of the biological properties of a mAb is of utmost importance and to achieve this, manufacturers rely on a reference standard, most often an in-house reference standard, throughout the product’s lifespan. Bioengineered mAbs which do not have naturally occurring similars are made available commercially in mass units, although their biological activity is exclusively defined on the basis of in-house proprietary reference standards.
Because of the rising number of mAb drug candidates and drugs in the market, the World Health Organization(WHO) has realized the requirement for global standardization of mAbs, to assure their safety, quality and efficacy. WHO in collaboration with regulatory authorities, manufacturers and other stakeholders, are developing WHO international standards (IS) for mAbs, that can be used for quality control and calibration of potency bioassays, as well as in-house reference standards. Furthermore, by defining the international unit (IU) of bioactivity, WHO IS for mAbs aid the assessment and management of mAb biologics utilized by different manufacturers and jurisdictions.


Recombinant mAbs are one of the fastest growing class of biotherapeutics and therefore WHO emphasizes on the need for establishing public or official reference standards for biopharmaceutical development. These reference materials are intended for the calibration of product potency and biological activity of in-house reference standards as well as national reference standards.
Owing to the complex nature and process-specific features of mAb drugs, it is implausible to develop a single international reference standard that can cover all the attributes of a product. Therefore, multiple attribute-specific reference standards are often required for analysis and process development.
Institutes such as NIST (US), NIBSC (UK) and regional pharmacopeial agencies are government recognized providers of metrological reference standards.
The NIST monoclonal antibody (NISTmAb) reference material, RM 8671, is intended for use in evaluating the performance of methods for determining physicochemical and biophysical attributes of monoclonal antibodies.
Further collaborative efforts between drug manufacturers, instrument manufacturers, regulatory authorities and standards organizations are necessary to develop new metrological reference standards that can supplement best industry practices to ensure product quality of mAb drugs.
The FDA industry guidance draft “Referencing Approved Drugs Products in ANDA Submissions” clearly states the difference between the reference standard and reference drugs for new generic drug applications (ANDA). According to the FDA guidance “A reference standard, which is selected by FDA, is the specific drug product that the ANDA applicant must use in conducting any in vivo bioequivalence testing”, generally this reference standard is the reference listed drug.
However, for the monoclonal antibodies and other biologic drugs, there is a
Therefore, it’s very difficult to obtain the desired reference standards for methods and assay development. In such a scenario the closest alternative is the approved reference drugs available on the market.
The approved monoclonal antibody drugs or other RLDs can serve as an alternative to reference standards.
Like reference standards, reference drugs have been thoroughly characterized and this information can serve as a valuable tool in assisting analytical scientists. Therefore, you can benchmark your methods (qualifying and validating) with the help of reference drugs.
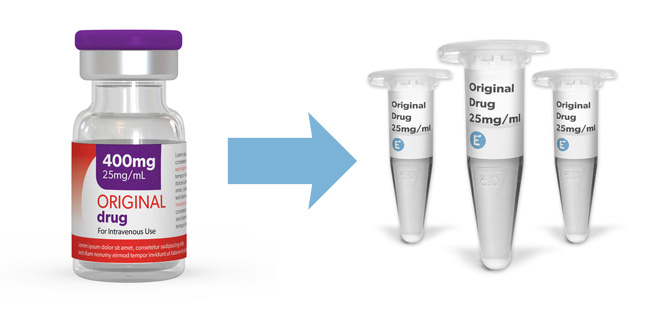
Evidentic supplies clinical-grade molecules from original reference drugs as research consumables. Our aliquots contain the original formulation of the commercial drug as we just repurpose the content of the original vial into a smaller and more convenient presentation. Check our portfolio and order today your desired molecules.